Prionové choroby jsou degenerativní onemocnění nervového systému, především mozku. Patří k nim scrapie u ovcí a koz, kuru a Creutzfeldt-Jakobova nemoc u lidí nebo bovinní spongiformní encefalopatie („nemoc šílených krav“) u skotu. Ta může být přenosná i na člověka, pokud konzumuje některé výrobky z nakažených zvířat.
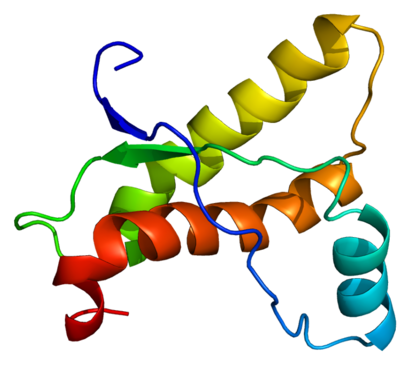
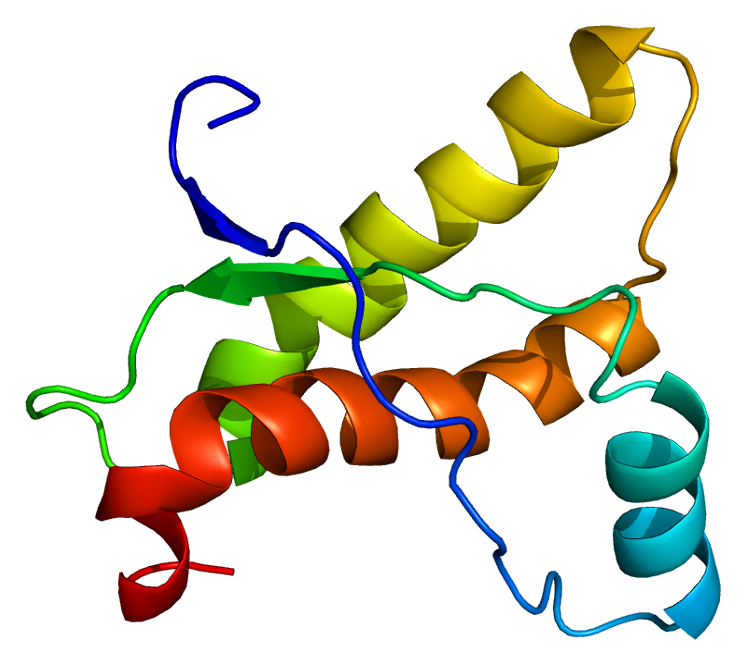
Původci těchto chorob jsou takzvané prionové proteiny. Jde o bílkoviny, které se v organizmu běžně vyskytují. Pokud mají jejich molekuly normální tvar, jsou neškodné. Prion se změněným („infekčním“) prostorovým uspořádáním ovšem vyvolává přeměnu normálních prionových molekul na „infekční“. Ty se pak shlukují, což vede k poškození nervové tkáně a propuknutí choroby.
Odlišné prostorové uspořádání molekuly má kromě shlukování také další důsledek – přílišnou odolnost prionu vůči trávicím enzymům, které by ho mohly rozštěpit na kratší řetězce aminokyselin (peptidy) a na jednotlivé aminokyseliny.
Nebezpečná forma prionu díky tomu projde agresivním prostředím žaludku. Následně se vstřebá ve střevě prostřednictvím bílých krvinek, které jsou součástí sliznice a tvoří zde imunitní bariéru, nebo jsou přítomny kvůli chronickému zánětu sliznice. Ani bílé krvinky však nejsou schopné prion strávit. Po jejich zániku se tak dostává do organismu, kde přispívá ke zvrhnutí normálních verzí proteinu do struktury způsobující potíže.